This is the second excercise.
mkdir -p kata/data
cd kata/data
screen -S kata
cp /data-shared/vcf_examples/luscinia_vars.vcf.gz .
< /data-shared/vcf_examples/luscinia_vars.vcf.gz zcat | egrep -o 'DP=[^;]*' | sed 's/DP=//' > g.csv
Now you have to run this command on your "home" machine.
Katerinka$ scp [email protected]:projects/kata/g.csv .
chrfun() { zcat | grep -v '^#' | grep $1| egrep -o 'DP=[^;]*' | sed 's/DP=//' > $2.csv ;}
$1 is the name of the chromosome, $2 is the under which I want to safe my file. Then, we can run the function on the data, we are interested in.
< /data-shared/vcf_examples/luscinia_vars_flags.vcf.gz chrfun '^chrZ\s' zet
scp [email protected]:projects/kata/zet.csv .
setwd("~/Desktop/PřF UK/UNIX"
datag <- read.csv("g.csv", col.names = "DP")
datag %>% ggplot(aes(DP)) + geom_histogram() + scale_y_log10()
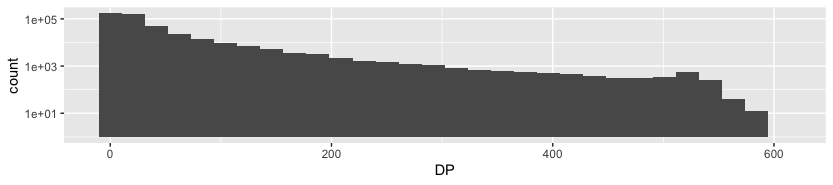
In this graph we can see how many reads (y) of a certain length (x) we can find after sequencing. There is a big varienty, taking into account, that y axis is logarithmical.
dataz <- read.csv("zet.csv", col.names = "DP")
dataz %>% ggplot(aes(DP)) + geom_histogram() + scale_y_log10()
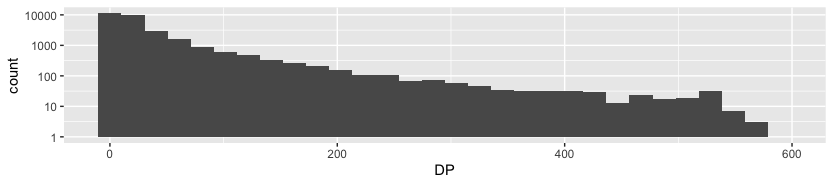
Since my solution shown above is plotting DP against its counts (so basically showing distribution of occurences of some DP), I decided to plot it against its loci, which can be quite useful (it helps us to know, which part of choromosome is being amplified most often).
Working in the same directory, let's remove headers and extract data only for the chromosome of interest
< /data-shared/vcf_examples/luscinia_vars_flags.vcf.gz zcat | grep -v '^#' | grep -e 'chr1\s' > data/nh.vcf
Now we can extract two columns into two separate steps and we have to save them as two separate files
(I tried to do it into one step, but it obviously doesn't work. I can't remove the column and then try to filter it...)
IN=data/nh.vcf <$IN cut -f2 > data/POS.tsv <$IN egrep -o 'DP=[^;]*' | sed 's/DP=//' > data/DP.tsv
paste data/POS.tsv data/DP.tsv > ch1.tsv
scp [email protected]:projects/kata/ch1.tsv .
setwd("~/Desktop/PřF UK/UNIX")
dat <- read_tsv("ch1.tsv", col_names=c("POS", "DP"))
dat %>% ggplot(aes(POS, DP)) + geom_line()
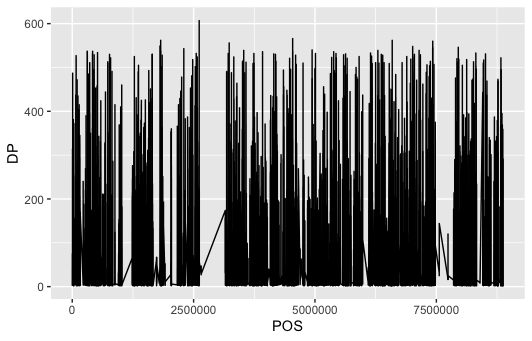
In the plot (having read depth on y and position of loci on x) we can see, that there are some gaps i reads – almost looks like some deletion event.
How will it look like for the whole genome? Same, just without column extraction according to the chromosome
< /data-shared/vcf_examples/luscinia_vars_flags.vcf.gz zcat | grep -v '^#' > data/nhg.vcf
IN=data/nhg.vcf <$IN cut -f2 > data/POSg.tsv <$IN egrep -o 'DP=[^;]*' | sed 's/DP=//' > data/DPg.tsv
paste data/POSg.tsv data/DPg.tsv > gg.tsv
scp [email protected]:projects/kata/gg.tsv .
datg <- read_tsv("gg.tsv", col_names=c("POS", "DP"))
dat %>% ggplot(aes(POS, DP)) + geom_line()
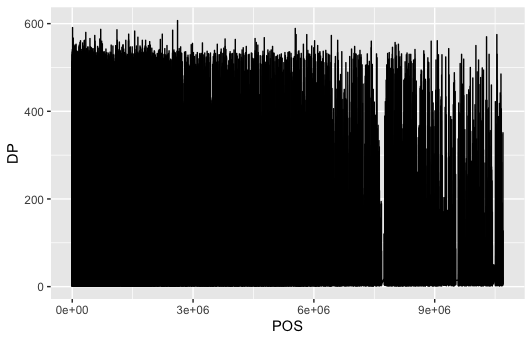
We can see, that the genome isn't covered in some portions.